Spatial averaging of the potential energy function facilitates the
search for the most stable configuration of a molecular system. Recently
some global optimization methods of this kind have been designed in the
literature that rely on physical phenomena such as diffusion, wave
function evolution in quantum mechanics, Smoluchowski dynamics,
evolution in temperature of canonical ensembles, etc. In the present
article we highlight the fact that all these methods, when applied to
the Gaussian distributions of an ensemble, represent special cases of a
set of differential equations involving the spatially averaged potential
energy. Their structure suggests that the nature's strategy to cope with
the global optimization is robust and differs only in the details in
particular applications. The strategy consists of going downhill of the
averaged potential energy, removing the barriers, and hunting for low
energy regions by a selective increasing of the spatial averaging. In
this study we explore the deformation of the potential rather than its
averaging. The deformation comes from scaling of atomic distances and
reduces the barriers even more effectively than the Gaussian averaging.
The position and widths of the Gaussian distribution evolve similarly to
the Gaussian density annealing (GDA), but we allow elliptical instead of
spherical Gaussians as well as branching of the single trajectory of the
system into multiple ones. When the temperature reaches 0 K, one has a
number of independent Gaussian distributions, each corresponding to a
structure and (usually low) energy of the system. The multiple elliptic-Gaussian
distance scaling method has been applied to clusters of argon atoms (N=5,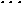 ,31),
a system serving usually as a benchmark domain. The method found the
global minima for all but three clusters (of very low energy). The
procedure is 20 or more times less expensive than the GDA one. ©
1997 John Wiley & Sons, Inc. J Comput Chem 18:
2040-2049, 1997 ,31),
a system serving usually as a benchmark domain. The method found the
global minima for all but three clusters (of very low energy). The
procedure is 20 or more times less expensive than the GDA one. ©
1997 John Wiley & Sons, Inc. J Comput Chem 18:
2040-2049, 1997 |